
Merge branch 'ceplas-dm-viktoria' into 'main'
Ceplas dm viktoria See merge request !2
Showing
- assays/SpeciesSelection/isa.assay.xlsx 0 additions, 0 deletionsassays/SpeciesSelection/isa.assay.xlsx
- assays/SpeciesSelection/protocols/.gitkeep 0 additions, 0 deletionsassays/SpeciesSelection/protocols/.gitkeep
- assays/SpeciesSelection/protocols/Cross-speciesPredictionModels.md 9 additions, 0 deletions...eciesSelection/protocols/Cross-speciesPredictionModels.md
- assays/SpeciesSelection/protocols/Intra-speciesModelsAndLeave-one-outCross-validation.md 5 additions, 0 deletions...ls/Intra-speciesModelsAndLeave-one-outCross-validation.md
- isa.investigation.xlsx 0 additions, 0 deletionsisa.investigation.xlsx
- studies/ArchitectureAndProposedModels/README.md 0 additions, 0 deletionsstudies/ArchitectureAndProposedModels/README.md
- studies/ArchitectureAndProposedModels/isa.study.xlsx 0 additions, 0 deletionsstudies/ArchitectureAndProposedModels/isa.study.xlsx
- studies/ArchitectureAndProposedModels/protocols/.gitkeep 0 additions, 0 deletionsstudies/ArchitectureAndProposedModels/protocols/.gitkeep
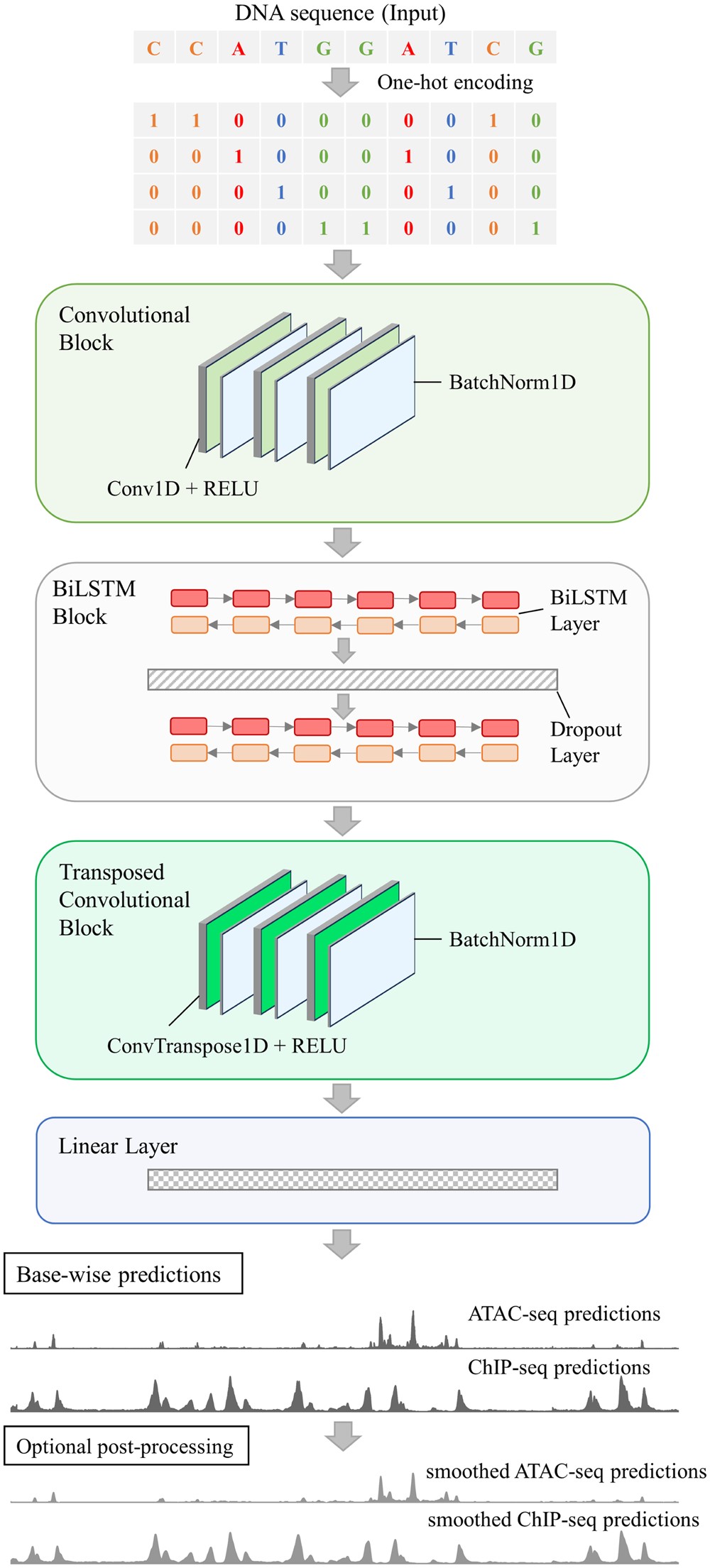
- studies/ArchitectureAndProposedModels/protocols/ArchitectureAndProposedModelsProtocol.md 11 additions, 0 deletions...Models/protocols/ArchitectureAndProposedModelsProtocol.md
- studies/ArchitectureAndProposedModels/resources/.gitkeep 0 additions, 0 deletionsstudies/ArchitectureAndProposedModels/resources/.gitkeep
- studies/ArchitectureAndProposedModels/resources/README.md 22 additions, 0 deletionsstudies/ArchitectureAndProposedModels/resources/README.md
- studies/ArchitectureAndProposedModels/resources/vbae074f2.jpeg 0 additions, 0 deletions...es/ArchitectureAndProposedModels/resources/vbae074f2.jpeg
- studies/Data/README.md 0 additions, 0 deletionsstudies/Data/README.md
- studies/Data/isa.study.xlsx 0 additions, 0 deletionsstudies/Data/isa.study.xlsx
- studies/Data/protocols/.gitkeep 0 additions, 0 deletionsstudies/Data/protocols/.gitkeep
- studies/Data/protocols/DataOverviewAndPreprocessing.md 5 additions, 0 deletionsstudies/Data/protocols/DataOverviewAndPreprocessing.md
- studies/Data/protocols/FilteringFlaggedSequences.md 3 additions, 0 deletionsstudies/Data/protocols/FilteringFlaggedSequences.md
- studies/Data/resources/.gitkeep 0 additions, 0 deletionsstudies/Data/resources/.gitkeep
- studies/Data/resources/README.md 37 additions, 0 deletionsstudies/Data/resources/README.md
- studies/Data/resources/vbae074f1.jpeg 0 additions, 0 deletionsstudies/Data/resources/vbae074f1.jpeg
assays/SpeciesSelection/isa.assay.xlsx
0 → 100644
File added
assays/SpeciesSelection/protocols/.gitkeep
0 → 100644
No preview for this file type
File added
{kind=link}
264 KiB
studies/Data/README.md
0 → 100644
studies/Data/isa.study.xlsx
0 → 100644
File added
studies/Data/protocols/.gitkeep
0 → 100644
studies/Data/resources/.gitkeep
0 → 100644
studies/Data/resources/README.md
0 → 100644
studies/Data/resources/vbae074f1.jpeg
0 → 100644
{kind=link}

434 KiB